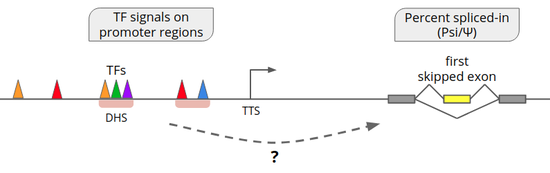
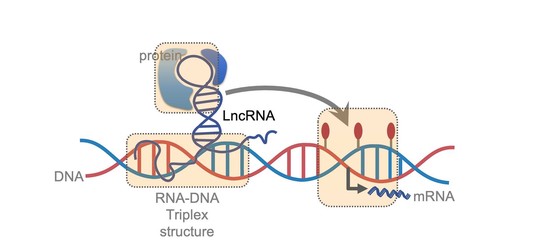
Bio-IT-Station
HK Tsai Lab of Bioinformatics
Institute of Information Science, Academia Sinica




Evolutionary Algorithm, Bioinformatics, Regulatory Mechanism, Metagenomics, Computational Biology
Statistical Computation, Machine Learning, Variables Selection in High Dimensional Data, Genomic